POLONEZ


Computational approaches to study protein-glycosaminoglycan interactions
- Funding Programme: POLONEZ from National Science Centre (Poland), European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 665778.
- Duration: 01.05.2017-30.04.2019 (project finished)
- Budget: 944 874 PLN (217 970 EUR)
- Project Leader: Sergey Samsonov
- Link to project description on NCN (Narodowe Centrum Nauki)
- Project website
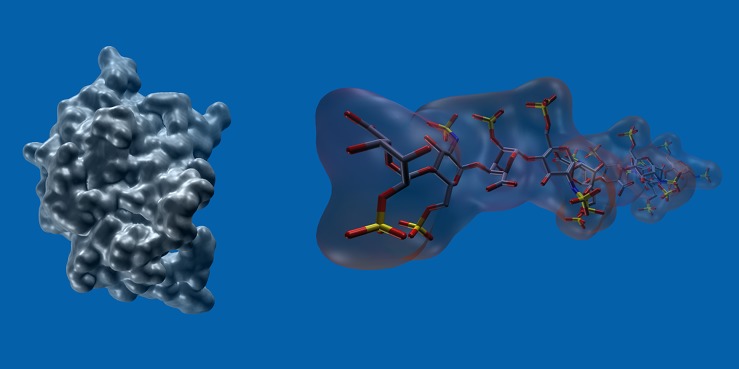
Glycosaminoglycans (GAGs) represent a class of linear anionic periodic polysaccharides comprised of disaccharide units containing an amino sugar and an uronic acid. They have different sulfation patterns contributing to their structural variety and molecular recognition. GAGs play a key role in many biological processes in the extracellular matrix via interactions with their protein targets such as chemokines and growth factors and so actively participate in cell signalling. Both naturally occuring and synthetic GAGs with modified sulfation patterns represent promising targets in artificial matrix engineering for potential applications in regenerative medicine. Despite the recently increased interest for protein-GAG systems within the theoretical chemistry community, there is still a lack of computational approaches developed specifically for GAGs because of many challenges originated from their molecular properties. First, in comparison to the available structural data for other biomolecules, the data on protein-GAG complexes are limited. Second, due to the highly charged nature of GAGs, appropriate treatment of electrostatics and solvent-mediated interactions for those systems is required. The periodic nature, linearity and fuctional groups disposition of GAGs render them difficult targets for distinguishing and scoring different binding poses when these molecules are bound at the protein surface. The fact that GAGs in the extracellular matrix are long polymers hinders the appliction of many classical computational tools developed for small molecules and make coarse-grained models attractive for those systems. Last but not least, while being inaccessible for convenient molecular dynamics (MD) time scales, pucker conformational space of uronic acid derivatives within GAGs can impose a crucial effect on their binding properties. Molecular docking approaches applied to GAG ligands also experience challenges due to 1. poor geometric complementarity in GAG binding site; 2. important role of water molecules for binding; 3. flexibility of long positively charged residue side-chains participating in GAG binding; 4. lack of specifically developed scoring schemes often requiring additional experimental data-based constraints.
In the proposed project, we would like to achieve two goals: 1. To carry out basic theoretical analysis of phosphorylated GAGs, which represent a novel and yet completely uncharacterized, both theoretically and experimentally, class of synthetically modified GAGs. These molecules are being produced and experimentally studied by our collaborators at the University of Leipzig (Germany) using mass-spectroscopy and nuclear magnetic resonanse (NMR) approaches. We will parameterize these molecules for all atomic and coarse-grained MD simulations, model the behaviour of series of GAGs with different phosphorylation pattern and various length to characterize their conformational space in details. The obtained properties will be compared with the corresponding properties of sulfated GAGs. Morever, in order to assist NMR spectra assignments, monomeric phosphorylated GAG building blocks will be simulated using Density Functional Theory approaches to calculated such NMR parameters as chemical shifts and J-coupling constants. 2. To computationally analyze protein-GAG interactions representing several protein targets: endostatin, PCPE-1 and cathepsins. The molecular-structural basis for endostatin and PCPE-1-GAG interactions will be studied in tight collaboration with the University of Lyon (France), where surface plasmon resonance (SPR) data on those interactions will be provided. For different cathepsins, ubiquitous proteases of the extracellular matrices, inhibition assays and SPR data with GAGs of various types are available from our collaborators from the University of Tours (France). Our previous work carried for cathepsins K and S, BMP-2, FGF, IL-8, SDF-1, sclerosin, OPG and other protein targets serves as a promising basis for application and further development of these approaches for endostatin-GAG, PCPE-1-GAG and cathepsin-GAG systems. The role of computational analysis, which will employ molecular docking, MD and free energy calculations approaches, is to gain the insights into the specificity of such interactions at atomic level. The project's success will be heavily dependent on the availability of powerful computational resources which are excellent at the hosting institution.
The results of the planned work will contribute to the general knowledge of protein-GAG molecular recognition and conformational properties of a novel class of phosphorylated GAG molecules, to understanding of appropriate computational strategies to handle these systems and will assist and complement the experimental data providing atomic details basis for underlying molecular mechanisms in protein-GAG interactions. The data obtained in this project will be useful for theoretical rationale in the further development of advanced and efficient approaches for tissue regeneration.
