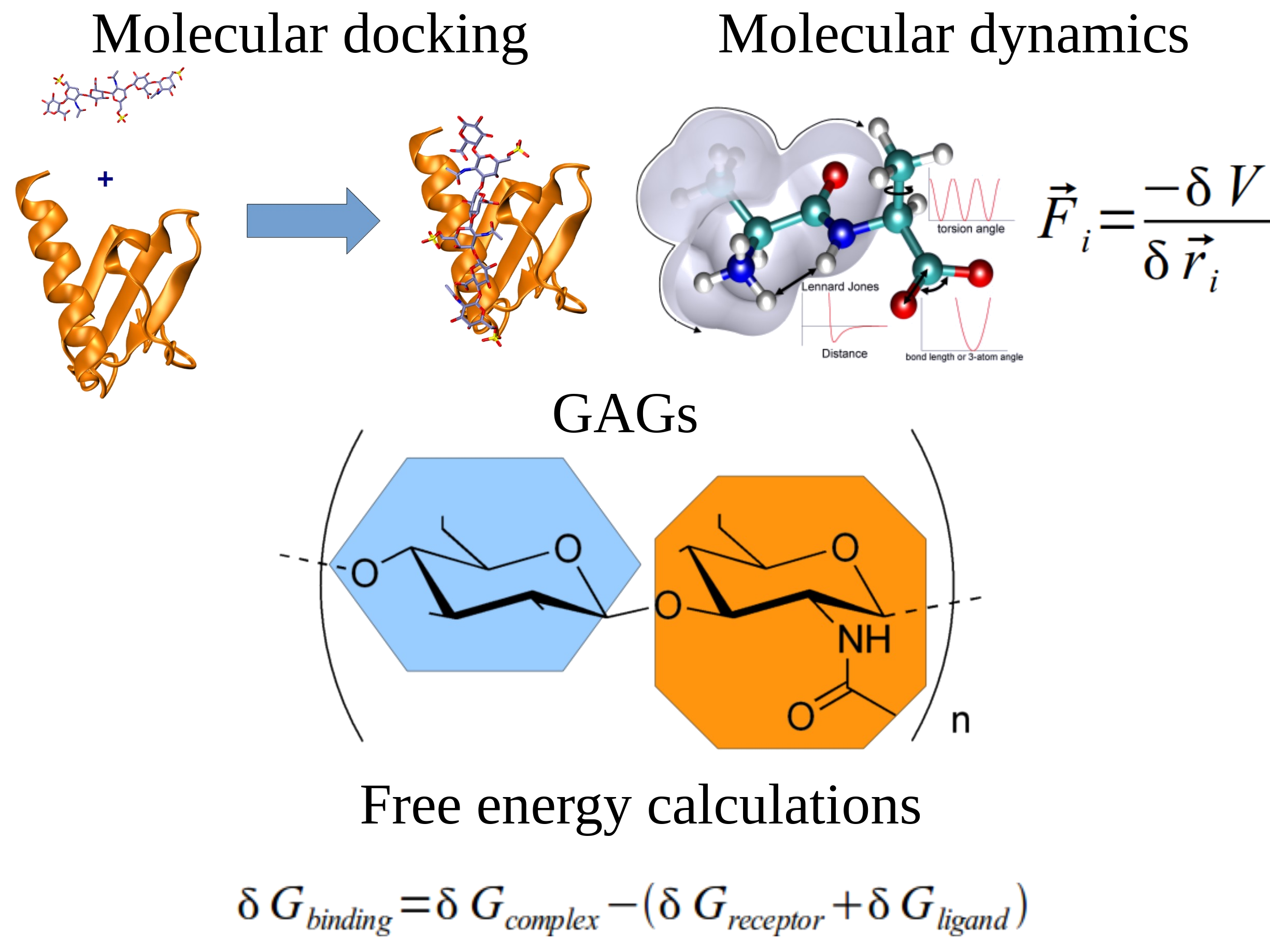
Research
Glycosaminoglycans (GAGs)
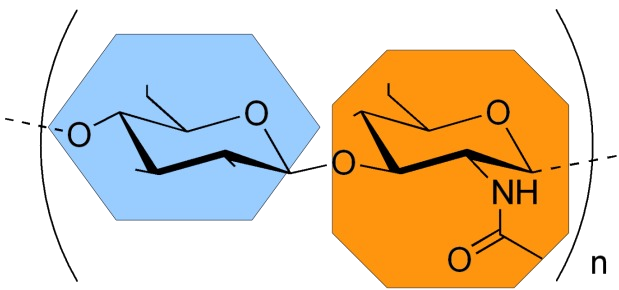
Glycosaminoglycans (GAGs) are linear periodic anionic polysaccharides playing a key role in the extracellular matrix via interactions with a manifold of proteins such as growth factors and chemokines and so representing promising targets in artificial matrix engineering for potential applications in regenerative medicine. Computationally, these molecules are challenging due to their high flexibility, particular sulfation pattern (“sulfation code”), importance of the solvent- and ion-mediated interactions, sugar ring conformational diversity, periodicity, multipose binding propensity and preference of binding to the long and flexible positively charged residues on the protein surface. In addition, when compared with other classes of molecules, there is a lack of specific computational approaches to effectively treat protein-GAG systems. In our group, we study different aspects of GAG-containing systems in silico and contribute to the development of GAG-specific computational approaches. Many ongoing projects in the group are carried out within collaborations with other experimental and theoretical groups.
Proteins-GAG interactions
GAGs are able to bind numerous proteins in the extracellular matrix and so modulate their activity. However, often the application of the experimental techniques alone does not allow to decipher the molecular mechanisms underlying these GAG-driven processes. We have established and tested a set of computational approaches based on molecular docking and molecular dynamics techniques, which could be successfully used for characterization of protein-GAG interactions in particular systems to complement, support and rationalize available experimental data. We have observed the dependence of protein-GAG interactions on GAG net sulfation, type and length. GAGs can be involved in the interactions of their protein targets with their receptors by different mechanisms: directly blocking receptor binding sites; shifting probability distributions in conformational ensemble of the protein influencing, in turn, the probability of binding the receptor; repulsing receptors via long- ranged electrostatic interactions; supporting interactions via formation of sandwich structures; predetermining the sequence of binding events in protein tertiary complexes.
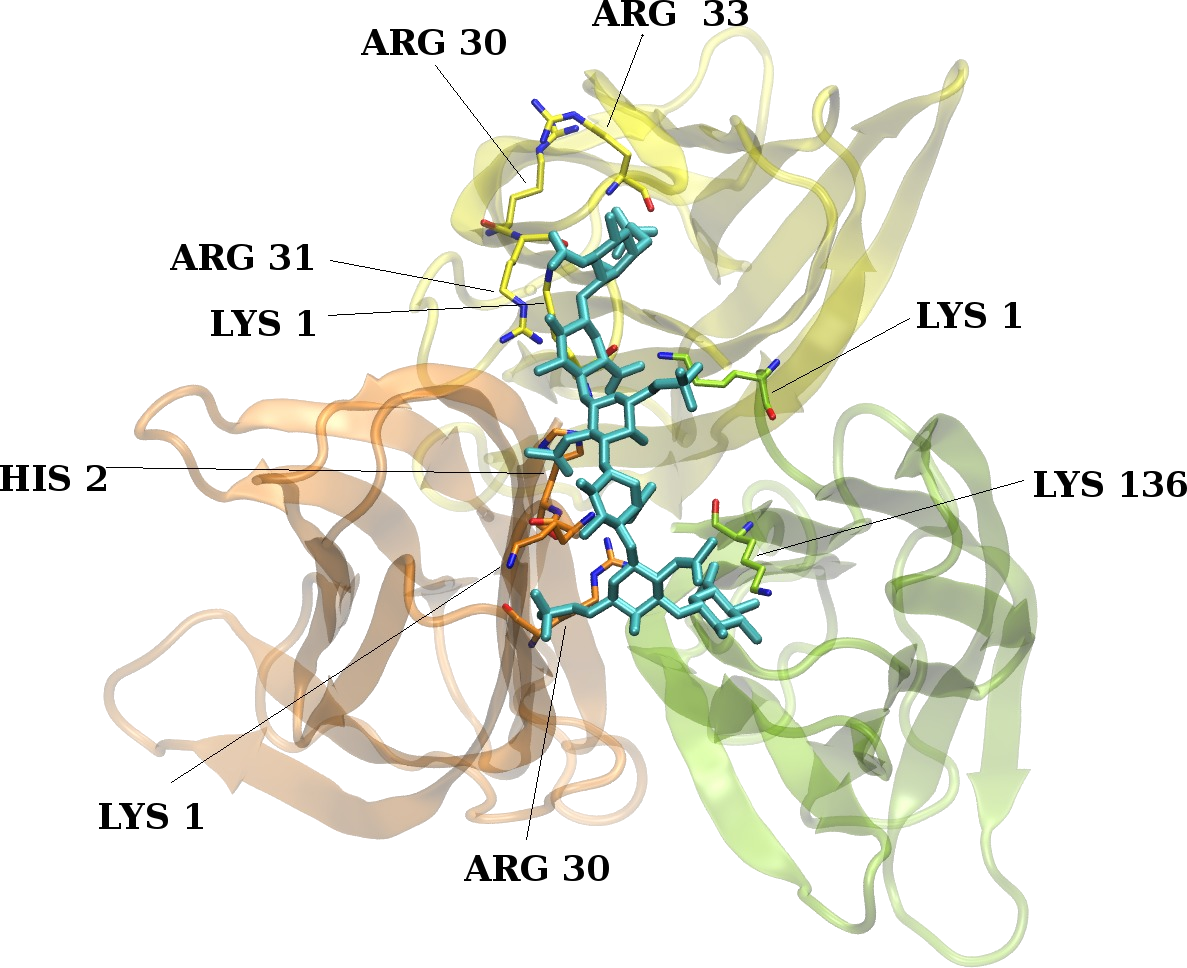
Peptide-GAG interactions
GAGs are known to have an impact on fibrillogenesis as well as to affect structural properties of antimicrobial peptides. Therefore, understanding of atomistic details of peptide-GAG interactions could have potentially high practical significance for the therapy of infectious diseases, including drug design and molecular diagnostics.
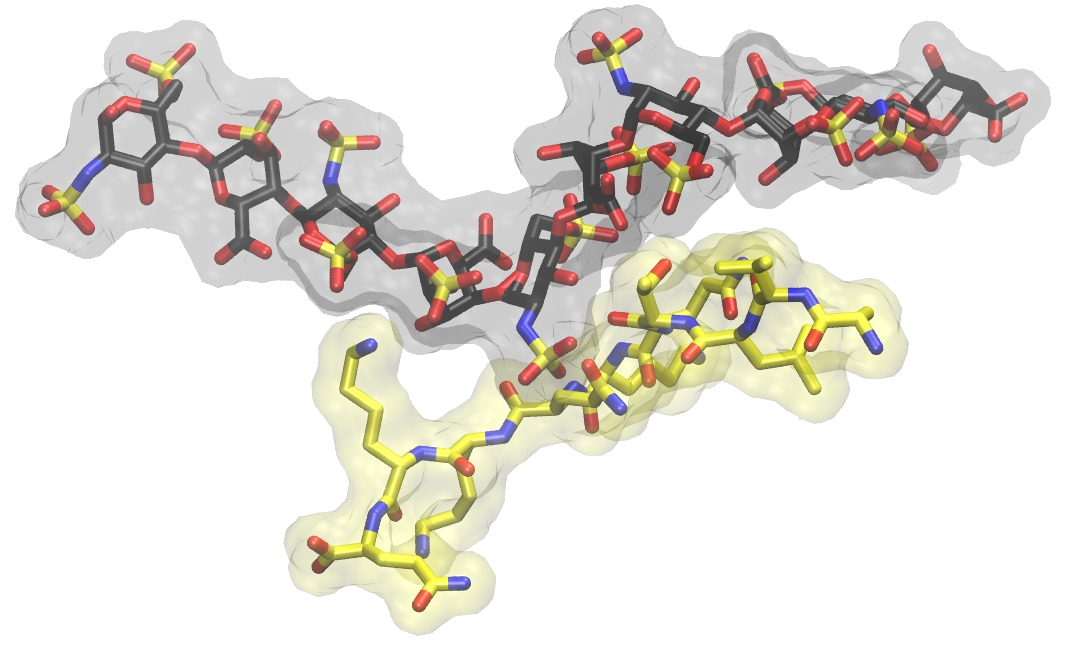
Small molecules-GAG interactions
GAGs reveal their potential ability of binding drug molecules including organic compounds originally targeted to the DNA minor groove or indirectly affecting gene expression suggesting a crucial role of GAGs in vascular diseases. Therefore, it is pivotal to unveil molecular mechanisms of GAG-small molecule non-covalent complex formation. We opt for obtaining and understanding the data contributing to the knowledge of molecular aspects of GAG interactions with drug molecules, which would potentially add to the comprehension of molecular pathways involved in their biological actions.
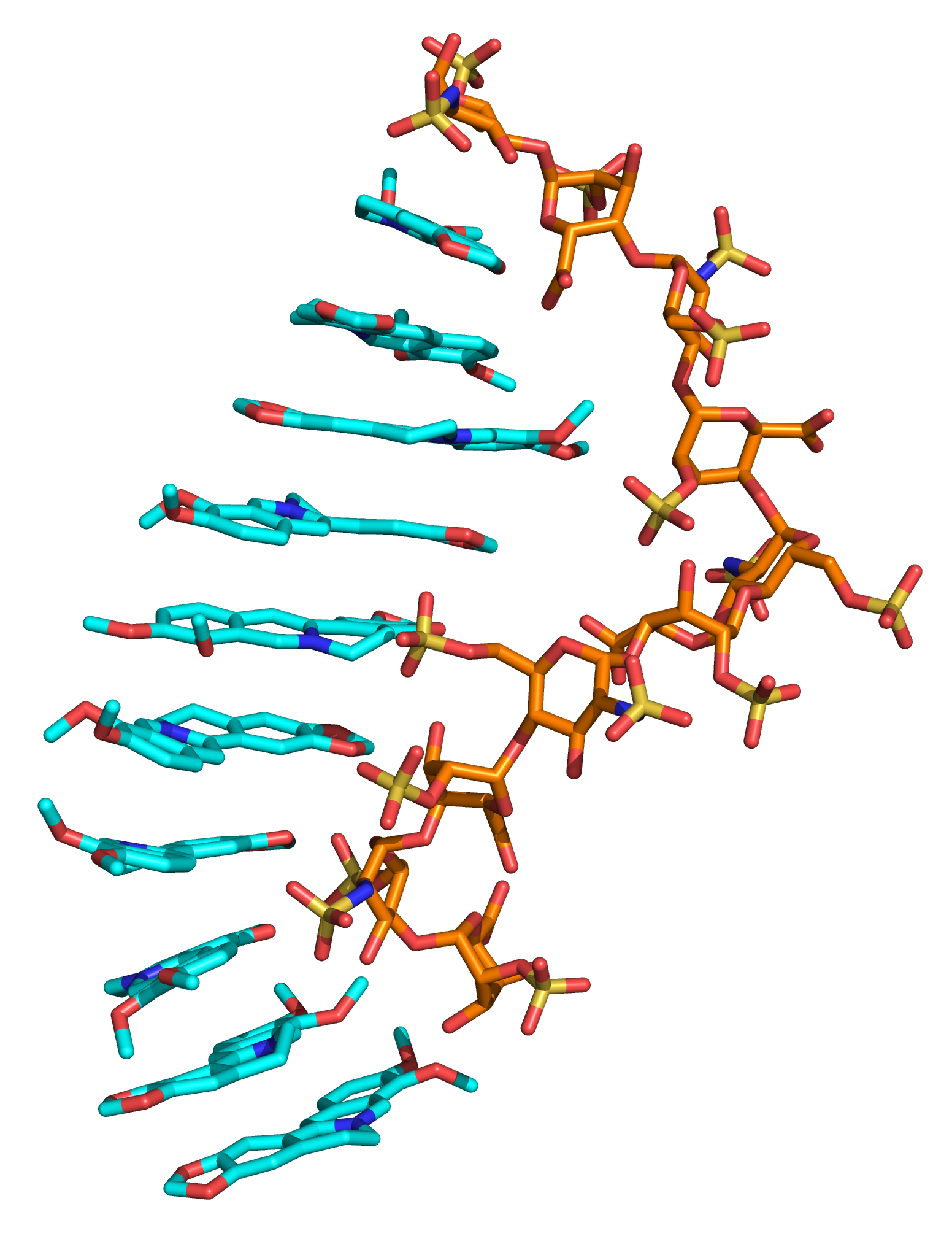
The role of ions
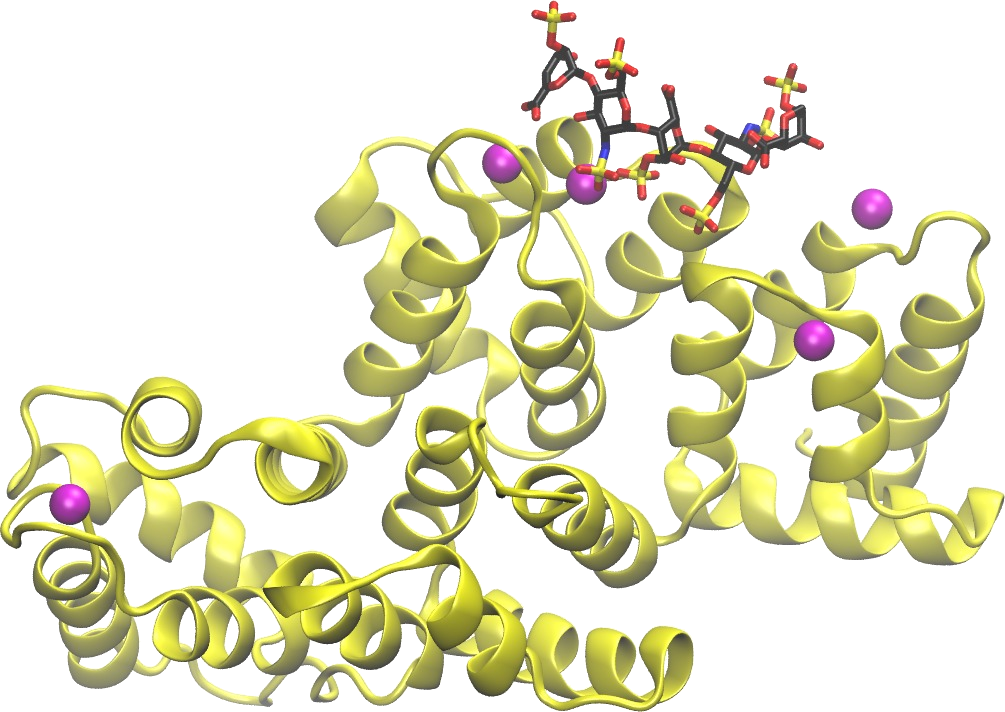
Specific binding and structural modifications caused by divalent ions on GAGs have been experimentally described for years. At the same time, there are just few attempts known up-to-date aimed at analyzing these interactions computationally. In addition to the effects on GAGs, divalent ions represent a decisive factor in many protein-GAG interactions, by their direct participation in the intermolecular interface or by changing the electrostatic properties of the protein surface or by influencing the conformational properties of GAGs. We aim to elucidate the potential mechanisms of interactions in these particularly challenging multicomponent systems made up of proteins, GAGs and ions.
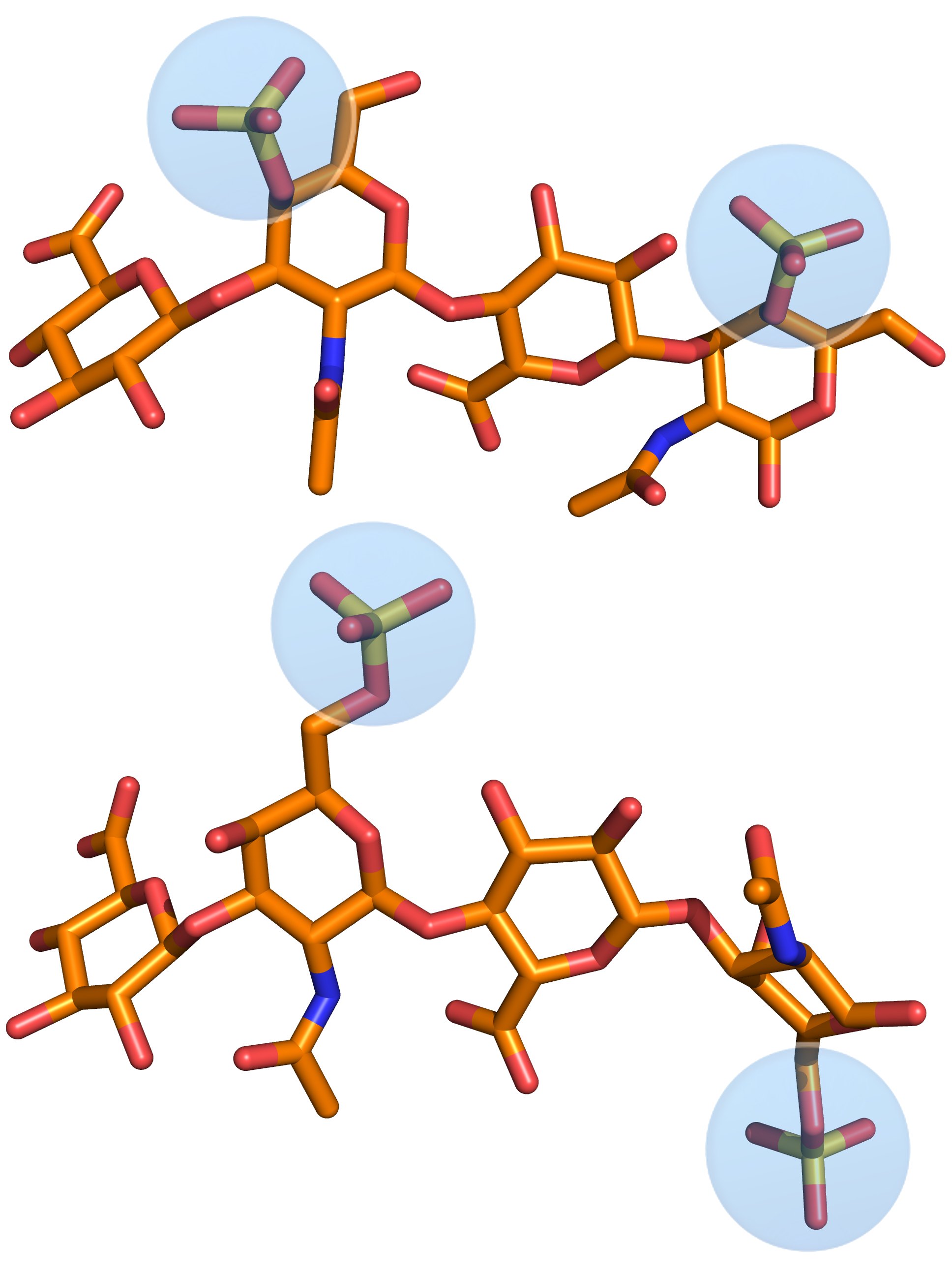
Specificity of GAG interactions
Among many challenges in GAG modeling, there is one question which is central in the field that remains unanswered: how specific are protein-GAG interactions? For several protein-GAG systems, it was shown that the alterations in the GAG type and sulfation pattern changes their protein binding capabilities specifically, whereas for others, it is demonstrated that the only parameter that affects the strength of such interactions is a GAG charge, and so these interactions are completely driven by electrostatics being, therefore, unspecific. However, there are no systematic studies dealing with the question of protein-GAG specificity. Moreover, the very concept of specificity often seems to be very unspecific itself. Together with the NMR research group of Prof. Huster (Leipzig University) we work on this topic to find out to which extent these highly charged molecules could be specific in terms of particular characteristics of their interactions with other biomacromolecules.
Coarse-grained modeling of GAG-containing systems
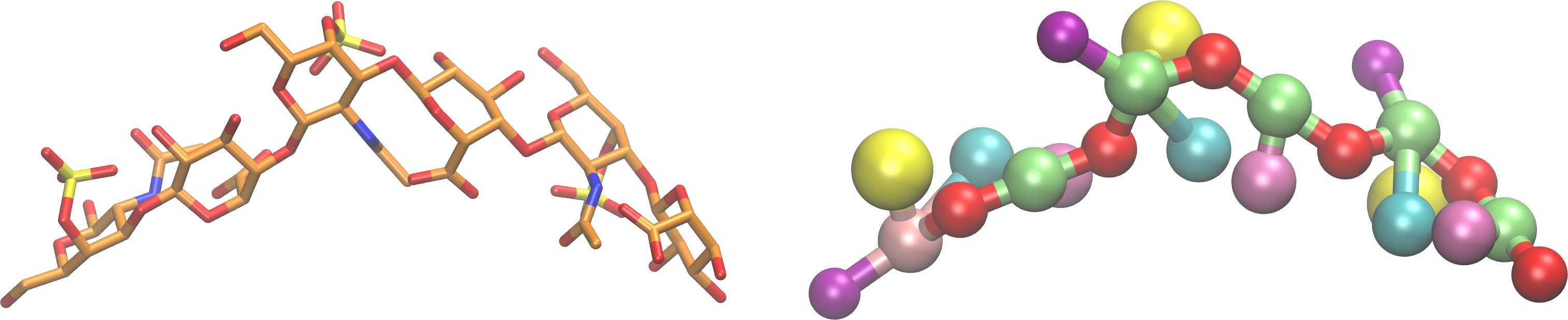
Many biologically relevant processes involving GAGs require modeling at time and distance scales essentially longer than the ones accessible by classical all atomic modeling. In these lines, the coarse-grained approach can bring essentially new type of data related to such processes as protein crowding or GAG-mediated protein/peptide structural enhancement. Our ongoing studies with the goal to implement GAG molecules within the framework of a coarse-grained modeling are performed in tight collaboration with Prof. Liwo (University of Gdansk) and his physics-based Unified Coarse-Grained Model force field. In case we succeed, this extension of the modeling scales will allow for principally new insights into the molecular mechanisms steered by the molecular interactions with GAGs within complex and versatile multicomponent systems.
Methodological aspects of GAG modeling and new tools development
More and more modeling approaches with the improved capabilities to treat GAG-containing systems have been appearing in the last decades. However, in comparison to small molecules, peptide, proteins and nucleic acids, rigorous analysis of these molecules is still in a high demand of further novel GAG-specific computational tools. In our group we are focused on modification and construction of new GAG-specific molecular docking approaches and free energy calculation schemes that would advance in terms of predictive power for these systems.